first-analysis
Philipp Bayer
2020-09-17
Last updated: 2020-12-11
Checks: 7 0
Knit directory: R_gene_analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2.9000). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200917) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 83dc49a. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Untracked files:
Untracked: data/Brec_R1.txt
Untracked: data/Brec_R2.txt
Untracked: data/CR15_R1.txt
Untracked: data/CR15_R2.txt
Untracked: data/CR_14_R1.txt
Untracked: data/CR_14_R2.txt
Untracked: data/KS_R1.txt
Untracked: data/KS_R2.txt
Untracked: data/NBS_PAV.txt.gz
Untracked: data/NLR_PAV_GD.txt
Untracked: data/NLR_PAV_GM.txt
Untracked: data/PAVs_newick.txt
Untracked: data/PPR1.txt
Untracked: data/PPR2.txt
Untracked: data/SNPs_newick.txt
Untracked: data/bac.txt
Untracked: data/brown.txt
Untracked: data/cy3.txt
Untracked: data/cy5.txt
Untracked: data/early.txt
Untracked: data/flowerings.txt
Untracked: data/foregeye.txt
Untracked: data/height.txt
Untracked: data/late.txt
Untracked: data/mature.txt
Untracked: data/motting.txt
Untracked: data/mvp.kin.bin
Untracked: data/mvp.kin.desc
Untracked: data/oil.txt
Untracked: data/pdh.txt
Untracked: data/protein.txt
Untracked: data/rust_tan.txt
Untracked: data/salt.txt
Untracked: data/seedq.txt
Untracked: data/seedweight.txt
Untracked: data/stem_termination.txt
Untracked: data/sudden.txt
Untracked: data/virus.txt
Untracked: data/yield.txt
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/first-analysis.Rmd) and HTML (docs/first-analysis.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 83dc49a | Philipp Bayer | 2020-12-11 | Trace deleted files |
| html | dae157b | Philipp Bayer | 2020-09-24 | Update of analysis |
| Rmd | 1b346e9 | Philipp Bayer | 2020-09-22 | More data! |
| html | 1b346e9 | Philipp Bayer | 2020-09-22 | More data! |
| html | 0777e1d | Philipp Bayer | 2020-09-22 | update |
| html | e6e9b9b | Philipp Bayer | 2020-09-21 | Build site. |
| Rmd | c1fbbf9 | Philipp Bayer | 2020-09-21 | wflow_publish("analysis/*") |
| Rmd | c71005a | Philipp Bayer | 2020-09-18 | lots changes |
| html | c71005a | Philipp Bayer | 2020-09-18 | lots changes |
| html | 7d33bac | Philipp Bayer | 2020-09-18 | Build site. |
| Rmd | 695db1e | Philipp Bayer | 2020-09-18 | wflow_publish(c(“analysis/eda.Rmd”, “analysis/first-analysis.Rmd”, |
| html | ca65f66 | Philipp Bayer | 2020-09-18 | Build site. |
| Rmd | efd6af1 | Philipp Bayer | 2020-09-18 | wflow_git_commit(all = T) |
| html | efd6af1 | Philipp Bayer | 2020-09-18 | wflow_git_commit(all = T) |
knitr::opts_chunk$set(warning = FALSE, message = FALSE)
library(tidyverse)
library(patchwork)
library(ggsci)
library(dabestr)
library(dabestr)
library(cowplot)
library(ggsignif)
theme_set(theme_cowplot())Introduction
npg_col = pal_npg("nrc")(9)
col_list <- c(`Wild-type`=npg_col[8],
Landrace = npg_col[3],
`Old cultivar`=npg_col[2],
`Modern cultivar`=npg_col[4])
pav_table <- read_tsv('./data/soybean_pan_pav.matrix_gene.txt.gz')NBS part
Let’s pull the NBS genes from the table
nbs <- read_tsv('./data/Lee.NBS.candidates.lst', col_names = c('Name', 'Class'))
# have to remove the .t1s
nbs$Name <- gsub('.t1','', nbs$Name)nbs_pav_table <- pav_table %>% filter(Individual %in% nbs$Name)names <- c()
percs <- c()
for (i in seq_along(nbs_pav_table)){
if ( i == 1) next
thisind <- colnames(nbs_pav_table)[i]
pavs <- nbs_pav_table[[i]]
perc <- sum(pavs) / length(pavs) * 100
names <- c(names, thisind)
percs <- c(percs, perc)
}
nbs_res_tibb <- new_tibble(list(names = names, percs = percs))OK what do these presence percentages look like?
ggplot(data=nbs_res_tibb, aes(x=percs)) + geom_histogram(bins=25) 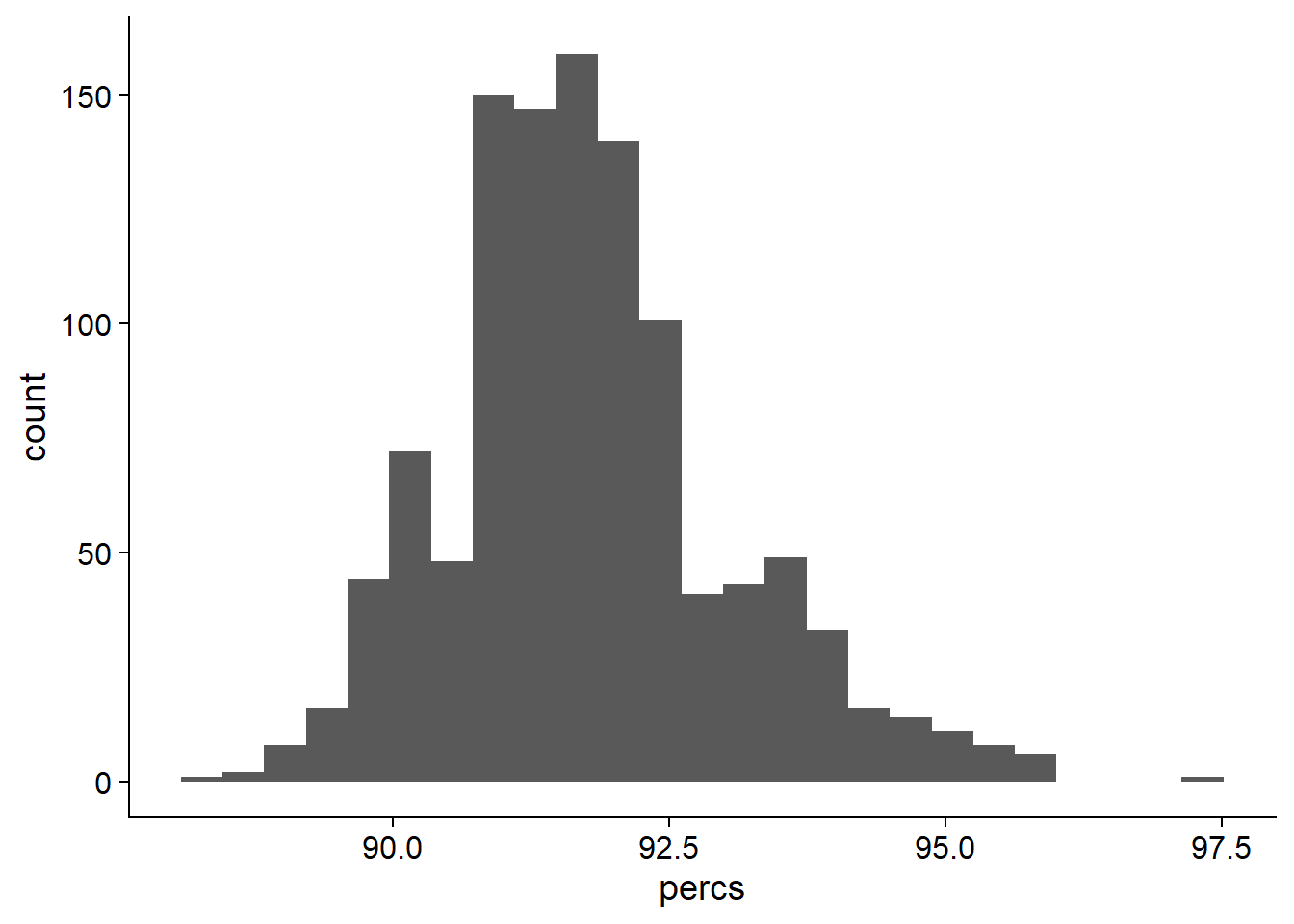
On average, 91.7701034% of NBS genes are present in each individual.
Now let’s join the table of presences to the four different types so we can group these numbers.
nbs_groups <- read_csv('./data/Table_of_cultivar_groups.csv')
nbs_joined_groups <- left_join(nbs_res_tibb, nbs_groups, by = c('names'='Data-storage-ID'))nbs_joined_groups$`Group in violin table` <- gsub('landrace', 'Landrace', nbs_joined_groups$`Group in violin table`)
nbs_joined_groups$`Group in violin table` <- gsub('Modern_cultivar', 'Modern cultivar', nbs_joined_groups$`Group in violin table`)
nbs_joined_groups$`Group in violin table` <- gsub('Old_cultivar', 'Old cultivar', nbs_joined_groups$`Group in violin table`)
nbs_joined_groups$`Group in violin table` <- factor(nbs_joined_groups$`Group in violin table`, levels=c(NA, 'Wild-type', 'Landrace', 'Old cultivar', 'Modern cultivar'))library(ggforce)
nbs_vio <- nbs_joined_groups %>% filter(`Group in violin table` != 'NA') %>%
ggplot(aes(y=percs, x=`Group in violin table`, fill=`Group in violin table`)) +
geom_violin(draw_quantiles = c(0.5)) +
geom_sina(alpha=0.5) +
geom_smooth(aes(group=1), method='glm') +
scale_fill_manual(values=col_list)+
guides(fill = FALSE) +
ylim(c(87, 100))
nbs_vio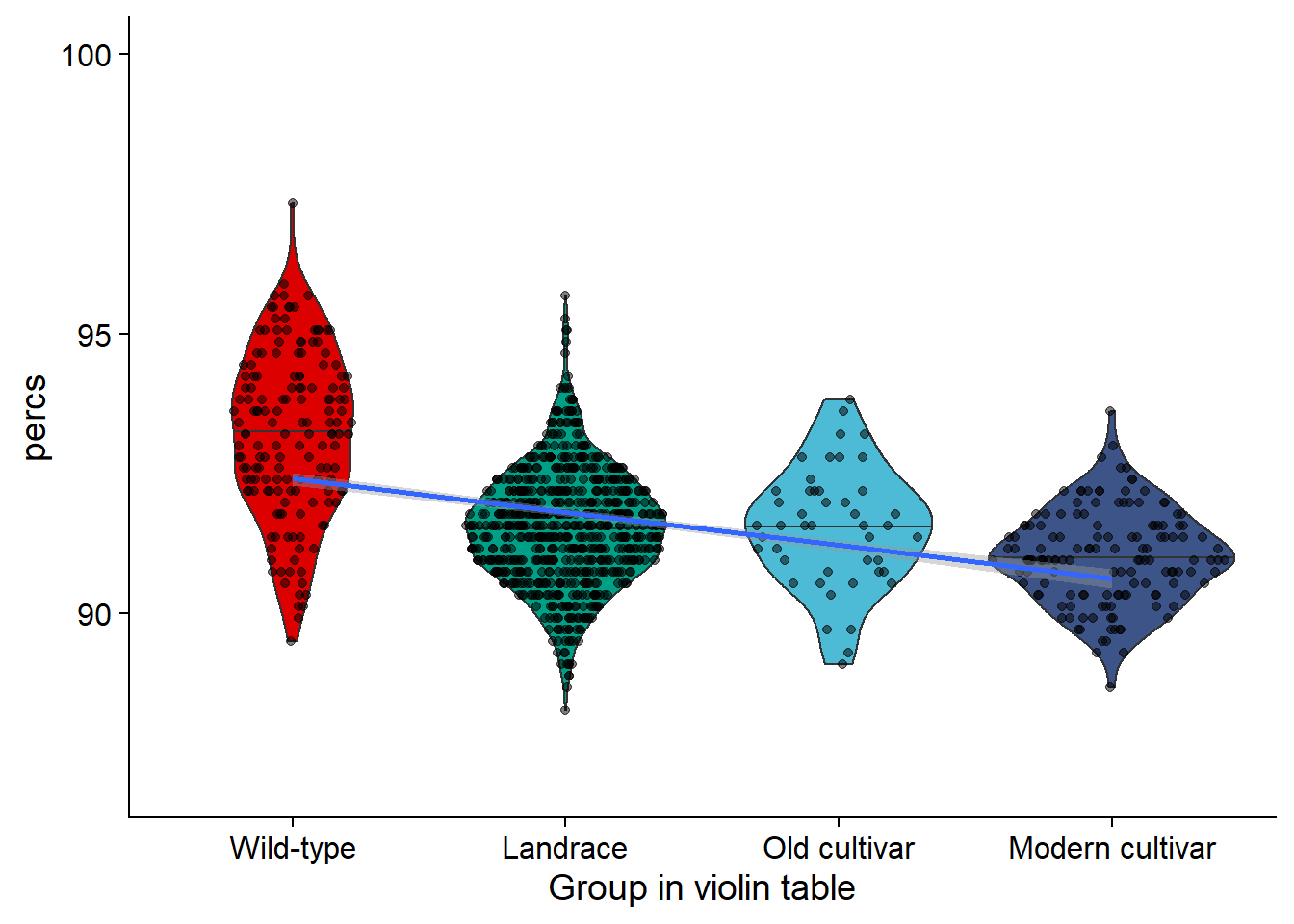
nbs_joined_groups %>% filter(`Group in violin table` != 'NA') %>%
ggplot(aes(y=percs, x=`Group in violin table`, fill=`Group in violin table`)) +
geom_smooth(aes(group=1), method='lm', se = FALSE) +
geom_jitter() +
scale_fill_manual(values=col_list)+
guides(fill = FALSE)# +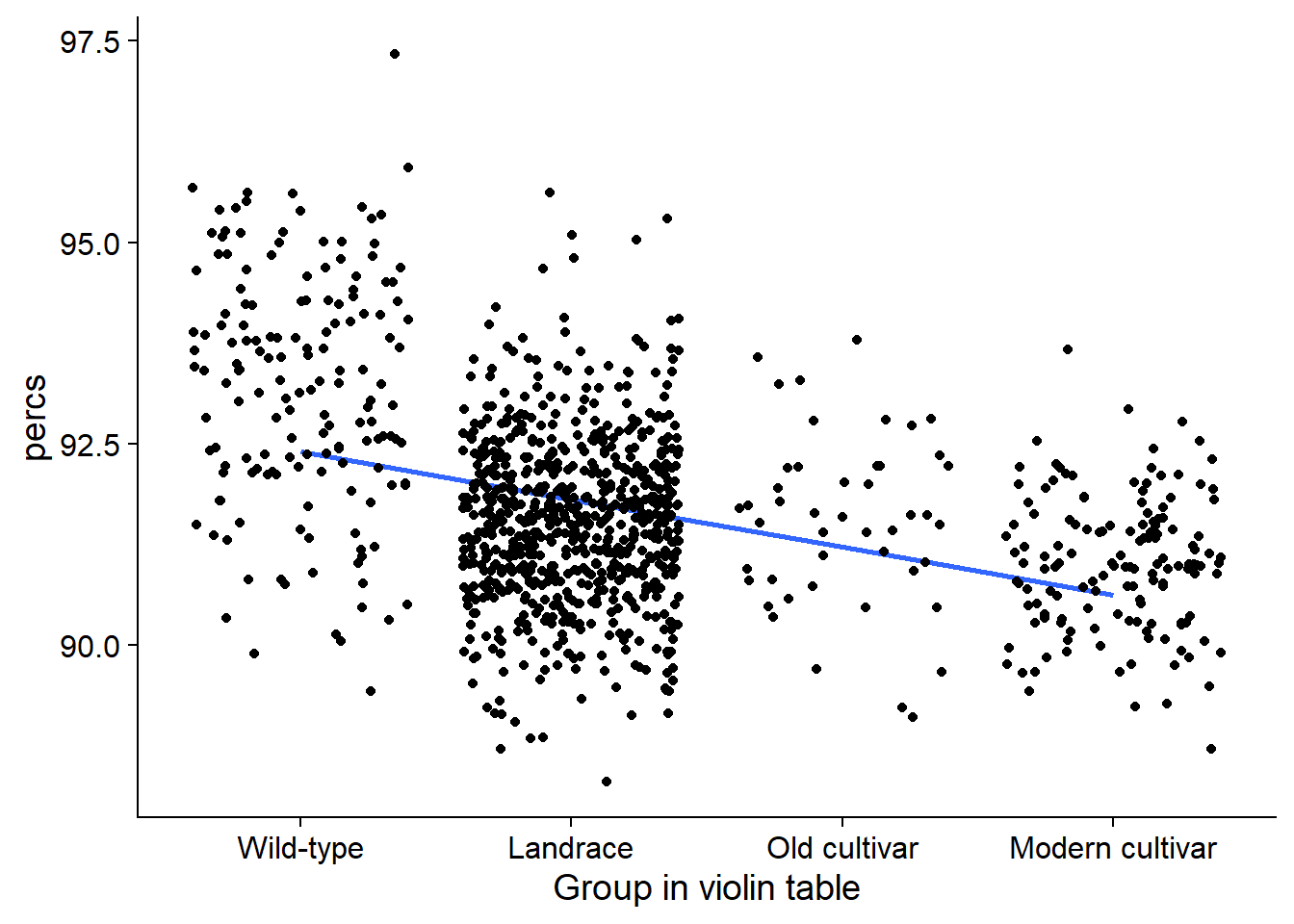
#ylim(c(0, 100))nbs_joined_groups %>% filter(!is.na(`PI-ID`)) %>%
group_by(`Group in violin table`) %>%
summarise(min_perc = min(percs),
max_perc = max(percs),
mean_perc = mean(percs),
median_perc = median(percs),
std_perc = sd(percs)) %>%
knitr::kable()| Group in violin table | min_perc | max_perc | mean_perc | median_perc | std_perc |
|---|---|---|---|---|---|
| Wild-type | 89.50617 | 97.32510 | 93.19939 | 93.20988 | 1.4754746 |
| Landrace | 88.27160 | 95.67901 | 91.54130 | 91.56379 | 1.0312082 |
| Old cultivar | 89.09465 | 93.82716 | 91.53695 | 91.56379 | 1.0701423 |
| Modern cultivar | 88.68313 | 93.62140 | 91.01125 | 90.94650 | 0.8329189 |
RLK part
Let’s do the same plot with RLKs
rlk <- read_tsv('./data/Lee.RLK.candidates.lst', col_names = c('Name', 'Class', 'Subtype'))
# have to remove the .t1s
rlk$Name <- gsub('.t1','', rlk$Name)rlk_pav_table <- pav_table %>% filter(Individual %in% rlk$Name)names <- c()
percs <- c()
for (i in seq_along(rlk_pav_table)){
if ( i == 1) next
thisind <- colnames(rlk_pav_table)[i]
pavs <- rlk_pav_table[[i]]
perc <- sum(pavs) / length(pavs) * 100
names <- c(names, thisind)
percs <- c(percs, perc)
}
rlk_res_tibb <- new_tibble(list(names = names, percs = percs))OK what do these presence percentages look like?
ggplot(data=rlk_res_tibb, aes(x=percs)) + geom_histogram(bins=25) 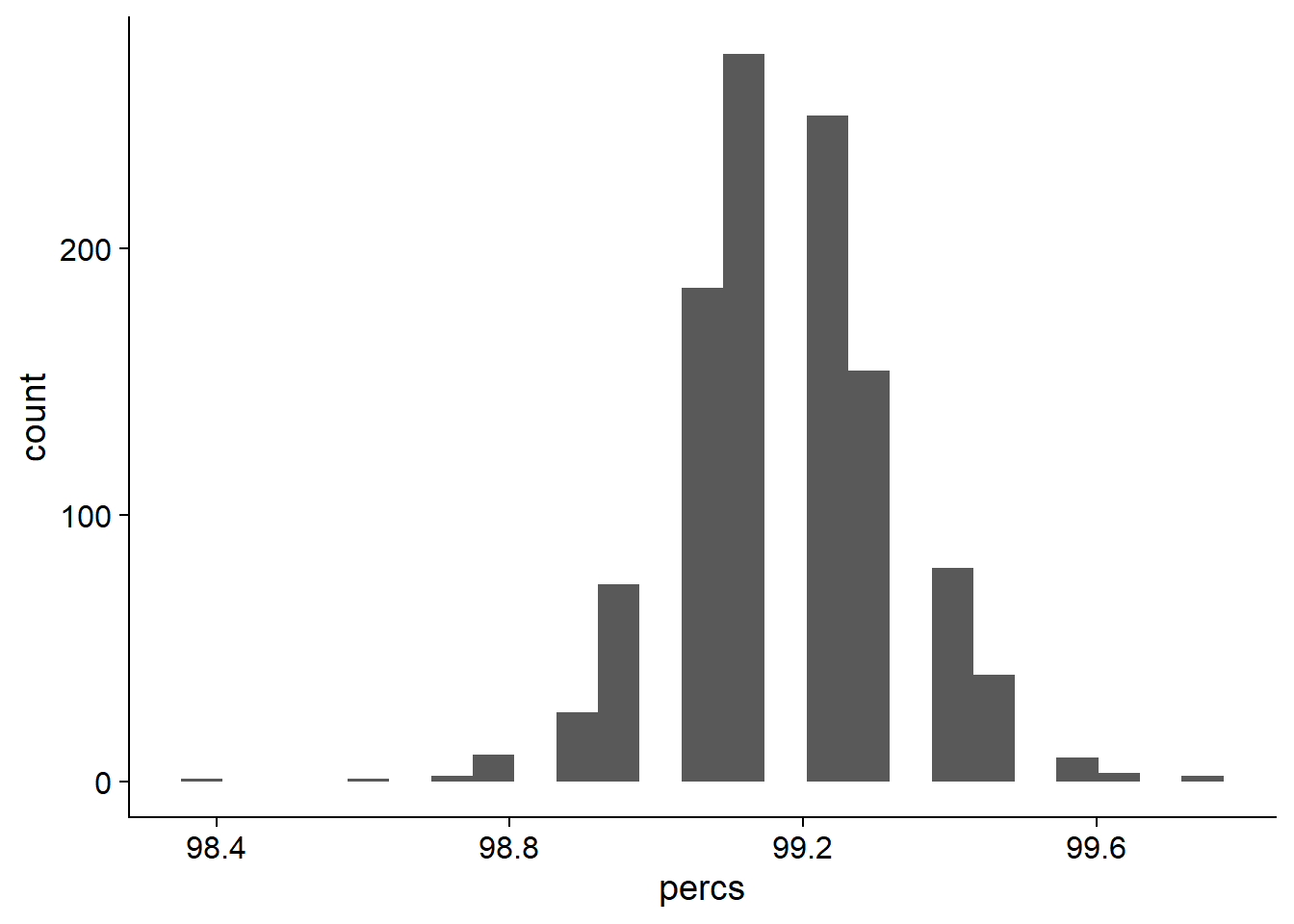
On average, 99.190418% of NBS genes are present in each individual.
Now let’s join the table of presences to the four different types so we can group these numbers.
groups <- read_csv('./data/Table_of_cultivar_groups.csv')
rlk_joined_groups <- left_join(rlk_res_tibb, groups, by = c('names'='Data-storage-ID'))rlk_joined_groups$`Group in violin table` <- gsub('landrace', 'Landrace', rlk_joined_groups$`Group in violin table`)
rlk_joined_groups$`Group in violin table` <- gsub('Modern_cultivar', 'Modern cultivar', rlk_joined_groups$`Group in violin table`)
rlk_joined_groups$`Group in violin table` <- gsub('Old_cultivar', 'Old cultivar', rlk_joined_groups$`Group in violin table`)
rlk_joined_groups$`Group in violin table` <- factor(rlk_joined_groups$`Group in violin table`, levels=c(NA, 'Wild-type', 'Landrace', 'Old cultivar', 'Modern cultivar'))rlk_vio <- rlk_joined_groups %>% filter(`Group in violin table` != 'NA') %>%
ggplot(aes(y=percs, x=`Group in violin table`, fill=`Group in violin table`)) +
geom_violin(draw_quantiles = c(0.5)) +
geom_sina(alpha=0.5) +
geom_smooth(aes(group=1), method='lm', se = FALSE) +
scale_fill_manual(values=col_list)+
guides(fill = FALSE)# +
#ylim(c(87, 100))
rlk_vio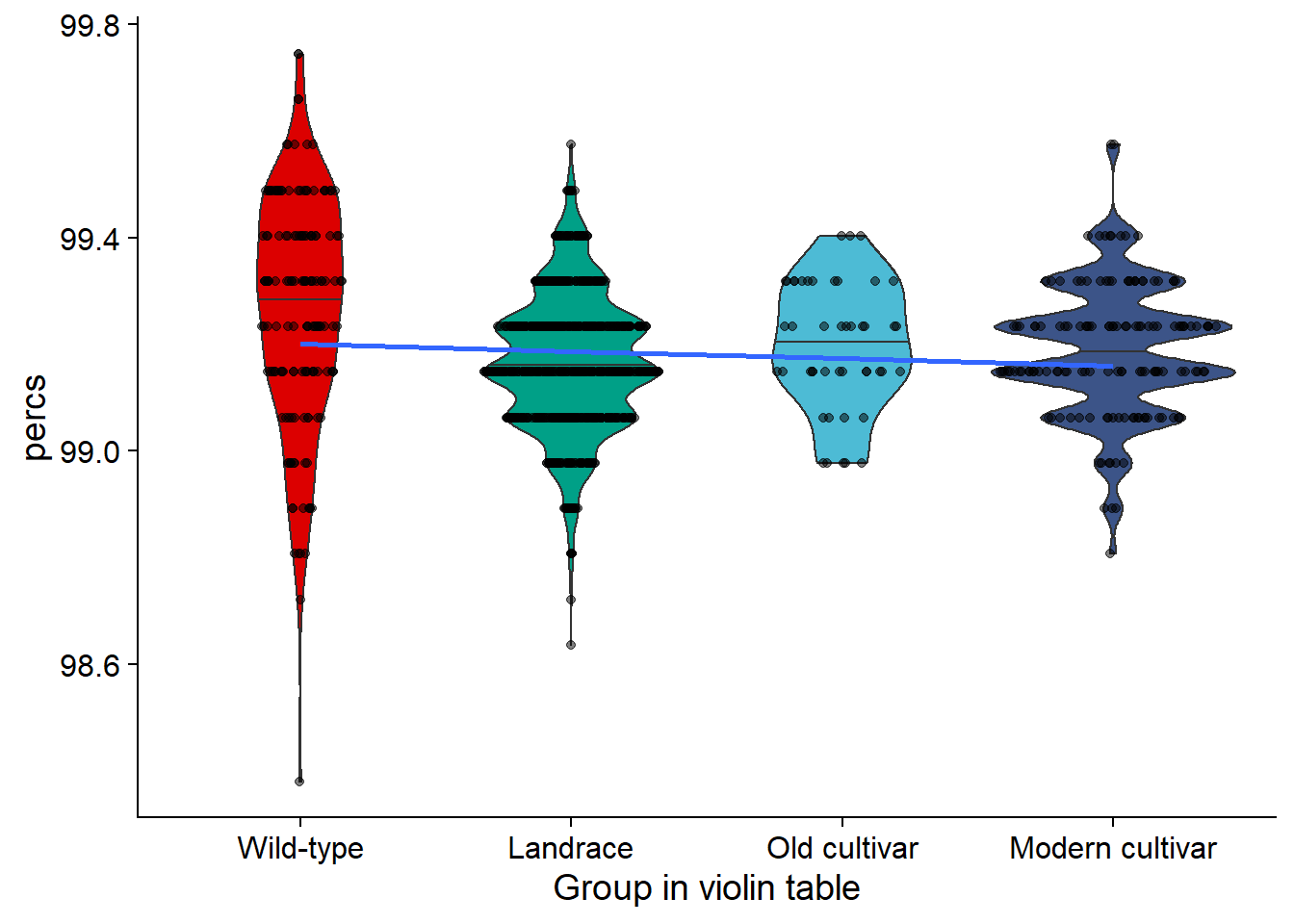
rlk_joined_groups %>% filter(!is.na(`PI-ID`)) %>%
group_by(`Group in violin table`) %>%
summarise(min_perc = min(percs),
max_perc = max(percs),
mean_perc = mean(percs),
median_perc = median(percs),
std_perc = sd(percs)) %>%
knitr::kable()| Group in violin table | min_perc | max_perc | mean_perc | median_perc | std_perc |
|---|---|---|---|---|---|
| Wild-type | 98.38022 | 99.74425 | 99.26314 | 99.31799 | 0.2177805 |
| Landrace | 98.63598 | 99.57374 | 99.16600 | 99.14749 | 0.1278145 |
| Old cultivar | 98.97698 | 99.40324 | 99.19752 | 99.23274 | 0.1199946 |
| Modern cultivar | 98.80648 | 99.57374 | 99.18922 | 99.14749 | 0.1255006 |
RLP part
And now with RLPs
rlp <- read_tsv('./data/Lee.RLP.candidates.lst', col_names = c('Name', 'Class', 'Subtype'))
# have to remove the .t1s
rlp$Name <- gsub('.t1','', rlp$Name)rlp_pav_table <- pav_table %>% filter(Individual %in% rlp$Name)names <- c()
percs <- c()
for (i in seq_along(rlp_pav_table)){
if ( i == 1) next
thisind <- colnames(rlp_pav_table)[i]
pavs <- rlp_pav_table[[i]]
perc <- sum(pavs) / length(pavs) * 100
names <- c(names, thisind)
percs <- c(percs, perc)
}
rlp_res_tibb <- new_tibble(list(names = names, percs = percs))OK what do these presence percentages look like?
ggplot(data=rlp_res_tibb, aes(x=percs)) + geom_histogram(bins=25) 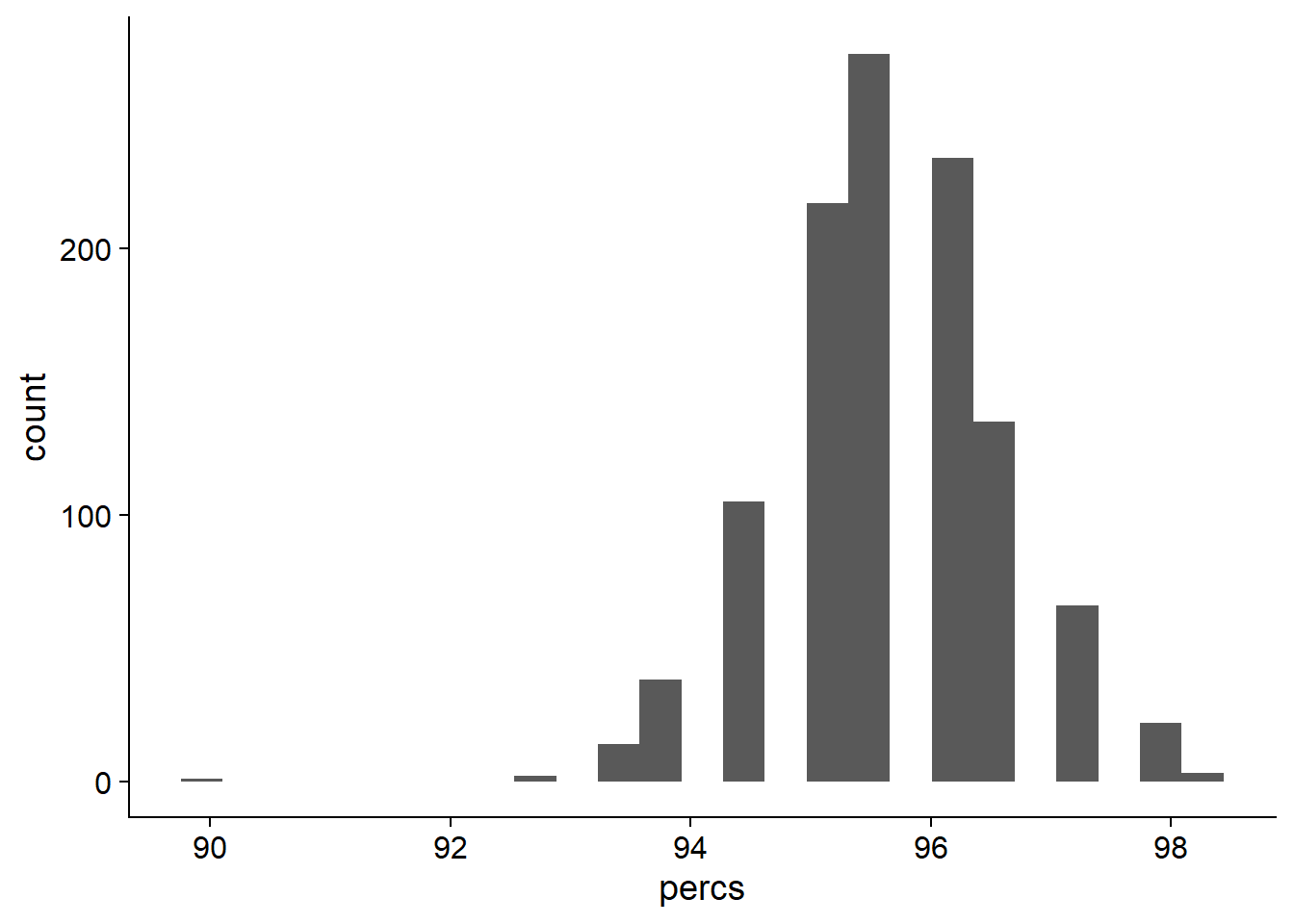
On average, 95.6496496% of NBS genes are present in each individual.
Now let’s join the table of presences to the four different types so we can group these numbers.
groups <- read_csv('./data/Table_of_cultivar_groups.csv')
rlp_joined_groups <- left_join(rlp_res_tibb, groups, by = c('names'='Data-storage-ID'))rlp_joined_groups$`Group in violin table` <- gsub('landrace', 'Landrace', rlp_joined_groups$`Group in violin table`)
rlp_joined_groups$`Group in violin table` <- gsub('Modern_cultivar', 'Modern cultivar', rlp_joined_groups$`Group in violin table`)
rlp_joined_groups$`Group in violin table` <- gsub('Old_cultivar', 'Old cultivar', rlp_joined_groups$`Group in violin table`)
rlp_joined_groups$`Group in violin table` <- factor(rlp_joined_groups$`Group in violin table`, levels=c(NA, 'Wild-type', 'Landrace', 'Old cultivar', 'Modern cultivar'))rlp_vio <- rlp_joined_groups %>% filter(`Group in violin table` != 'NA') %>%
ggplot(aes(y=percs, x=`Group in violin table`, fill=`Group in violin table`)) +
geom_violin(draw_quantiles = c(0.5)) +
geom_sina(alpha=0.5) +
geom_smooth(aes(group=1), method='lm', se = FALSE) +
scale_fill_manual(values=col_list)+
guides(fill = FALSE) +
ylim(c(87, 100))
rlp_vio
rlp_joined_groups %>% filter(`Group in violin table` != 'NA') %>%
ggplot(aes(y=percs, x=`Group in violin table`, fill=`Group in violin table`)) +
geom_jitter() +
#geom_sina(alpha=0.5) +
scale_fill_manual(values=col_list)+
guides(fill = FALSE) +
ylim(c(87, 100))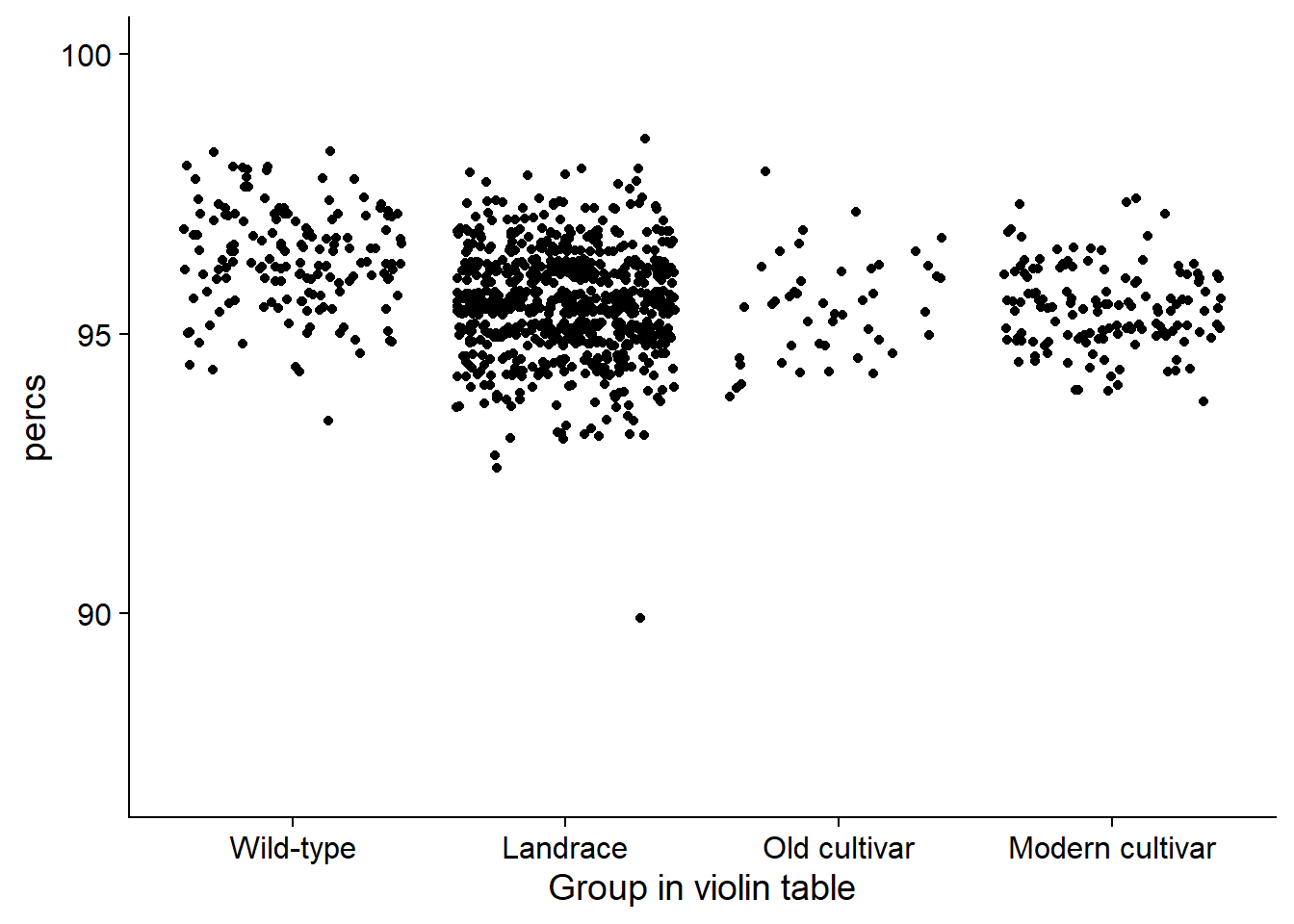
rlp_joined_groups %>% filter(!is.na(`PI-ID`)) %>%
group_by(`Group in violin table`) %>%
summarise(min_perc = min(percs),
max_perc = max(percs),
mean_perc = mean(percs),
median_perc = median(percs),
std_perc = sd(percs)) %>%
knitr::kable()| Group in violin table | min_perc | max_perc | mean_perc | median_perc | std_perc |
|---|---|---|---|---|---|
| Wild-type | 93.33333 | 98.33333 | 96.34112 | 96.11111 | 0.8985510 |
| Landrace | 90.00000 | 98.33333 | 95.53711 | 95.55556 | 0.9230701 |
| Old cultivar | 93.88889 | 97.77778 | 95.45894 | 95.55556 | 0.9019439 |
| Modern cultivar | 93.88889 | 97.22222 | 95.44678 | 95.55556 | 0.7169931 |
Plotting together
nbs_vio + rlk_vio + rlp_vio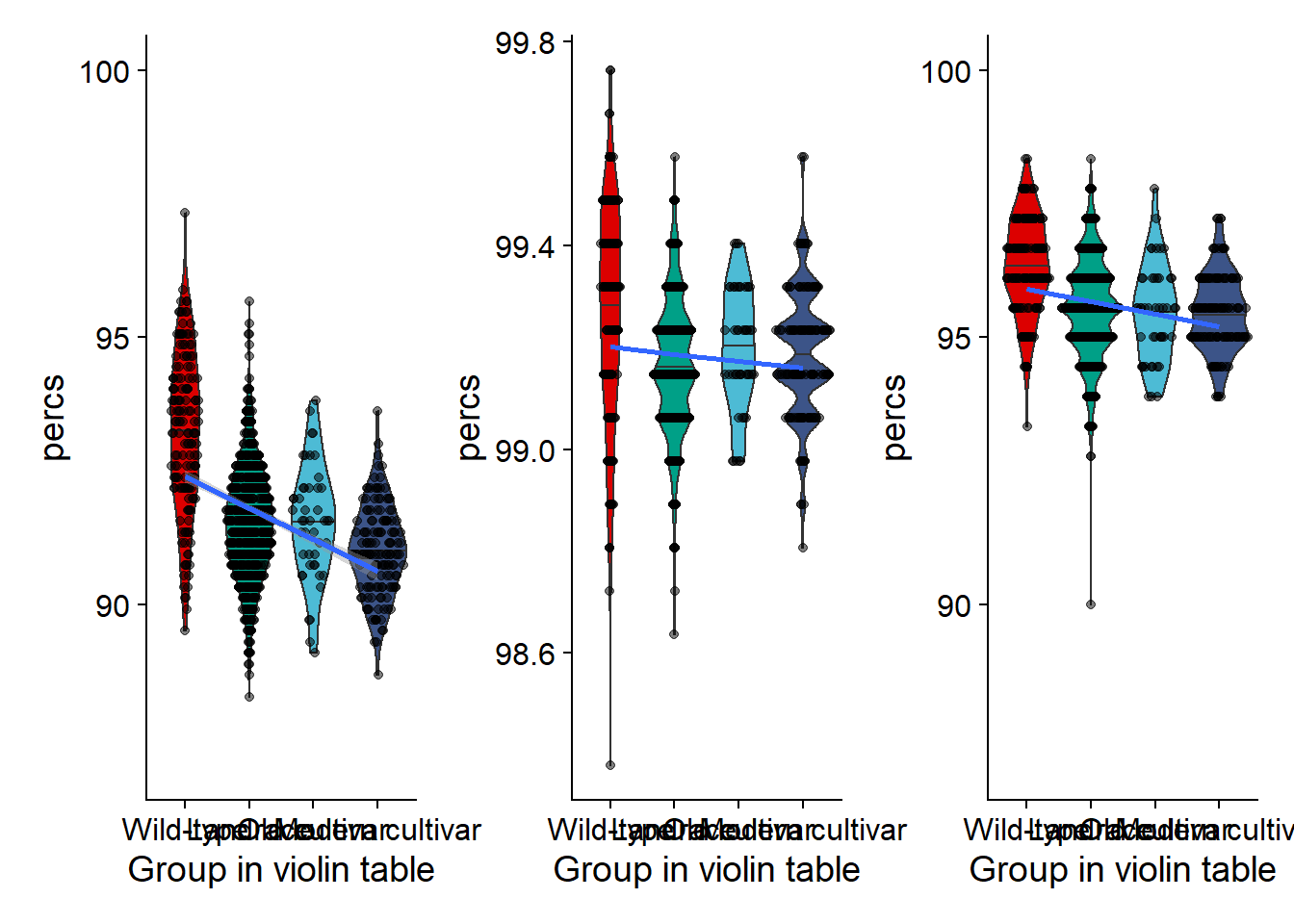
Stats - Dabayes
I want to know whether the groups are statistically significantly different. First let’s use dabestr
NBS
Let’s run dabestr first:
nbs_multi.two.group.unpaired <-
nbs_joined_groups %>% filter(!is.na(`PI-ID`)) %>%
dabest(`Group in violin table`, percs,
idx = list(c("Wild-type", "Landrace"),
c('Old cultivar', 'Modern cultivar')),
paired = FALSE)
nbs_multi.two.group.unpaireddabestr (Data Analysis with Bootstrap Estimation in R) v0.3.0
=============================================================
Good morning!
The current time is 11:04 AM on Friday December 11, 2020.
Dataset : .
The first five rows are:
# A tibble: 5 x 4
names percs `PI-ID` `Group in violin table`
<chr> <dbl> <chr> <fct>
1 AB-01 91.6 PI458020 Landrace
2 AB-02 93.4 PI603713 Landrace
3 DT2000 92.0 PI635999 Modern cultivar
4 For 92.2 PI548645 Modern cultivar
5 HN001 92.2 PI518664 Modern cultivar
X Variable : Group in violin table
Y Variable : percs
Effect sizes(s) will be computed for:
1. Landrace minus Wild-type
2. Modern cultivar minus Old cultivarnbs_multi.two.group.unpaired.meandiff <- mean_diff(nbs_multi.two.group.unpaired)
nbs_multi.two.group.unpaired.meandiffdabestr (Data Analysis with Bootstrap Estimation in R) v0.3.0
=============================================================
Good morning!
The current time is 11:04 AM on Friday December 11, 2020.
Dataset : .
X Variable : Group in violin table
Y Variable : percs
Unpaired mean difference of Landrace (n = 723) minus Wild-type (n = 157)
-1.66 [95CI -1.9; -1.41]
Unpaired mean difference of Modern cultivar (n = 143) minus Old cultivar (n = 46)
-0.526 [95CI -0.875; -0.2]
5000 bootstrap resamples.
All confidence intervals are bias-corrected and accelerated.plot(nbs_multi.two.group.unpaired.meandiff, color.column=`Group in violin table`,
rawplot.ylabel = 'Presence (%)', show.legend=FALSE)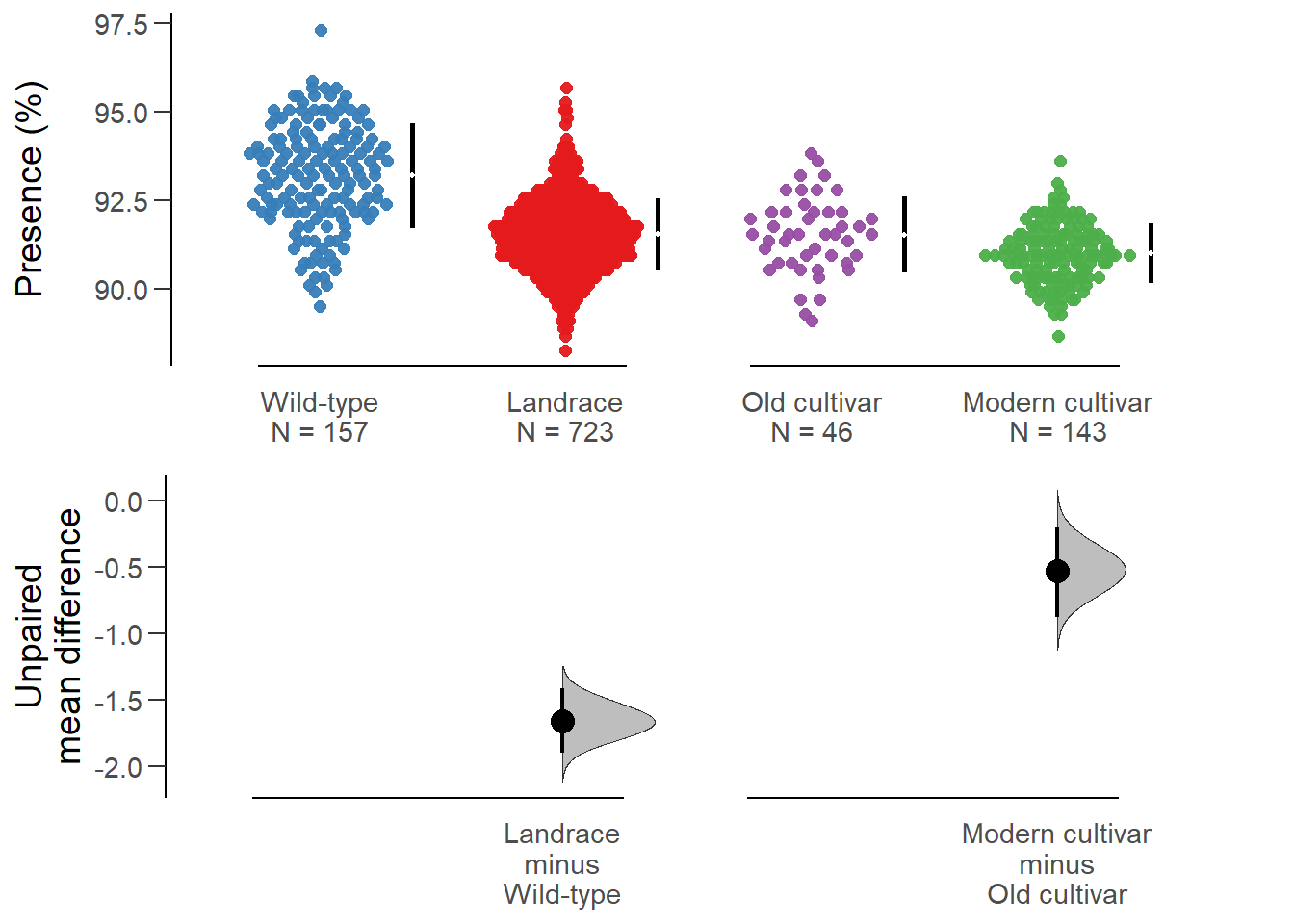
RLK
rlk_multi.two.group.unpaired <-
rlk_joined_groups %>% filter(!is.na(`PI-ID`)) %>%
dabest(`Group in violin table`, percs,
idx = list(c("Wild-type", "Landrace"),
c('Old cultivar', 'Modern cultivar')),
paired = FALSE)
rlk_multi.two.group.unpaireddabestr (Data Analysis with Bootstrap Estimation in R) v0.3.0
=============================================================
Good morning!
The current time is 11:04 AM on Friday December 11, 2020.
Dataset : .
The first five rows are:
# A tibble: 5 x 4
names percs `PI-ID` `Group in violin table`
<chr> <dbl> <chr> <fct>
1 AB-01 99.5 PI458020 Landrace
2 AB-02 99.1 PI603713 Landrace
3 DT2000 99.3 PI635999 Modern cultivar
4 For 99.1 PI548645 Modern cultivar
5 HN001 99.1 PI518664 Modern cultivar
X Variable : Group in violin table
Y Variable : percs
Effect sizes(s) will be computed for:
1. Landrace minus Wild-type
2. Modern cultivar minus Old cultivarrlk_multi.two.group.unpaired.meandiff <- mean_diff(rlk_multi.two.group.unpaired)
rlk_multi.two.group.unpaired.meandiffdabestr (Data Analysis with Bootstrap Estimation in R) v0.3.0
=============================================================
Good morning!
The current time is 11:04 AM on Friday December 11, 2020.
Dataset : .
X Variable : Group in violin table
Y Variable : percs
Unpaired mean difference of Landrace (n = 723) minus Wild-type (n = 157)
-0.0971 [95CI -0.132; -0.0611]
Unpaired mean difference of Modern cultivar (n = 143) minus Old cultivar (n = 46)
-0.00831 [95CI -0.0479; 0.0308]
5000 bootstrap resamples.
All confidence intervals are bias-corrected and accelerated.plot(rlk_multi.two.group.unpaired.meandiff, color.column=`Group in violin table`,
rawplot.ylabel = 'Presence (%)', show.legend=FALSE)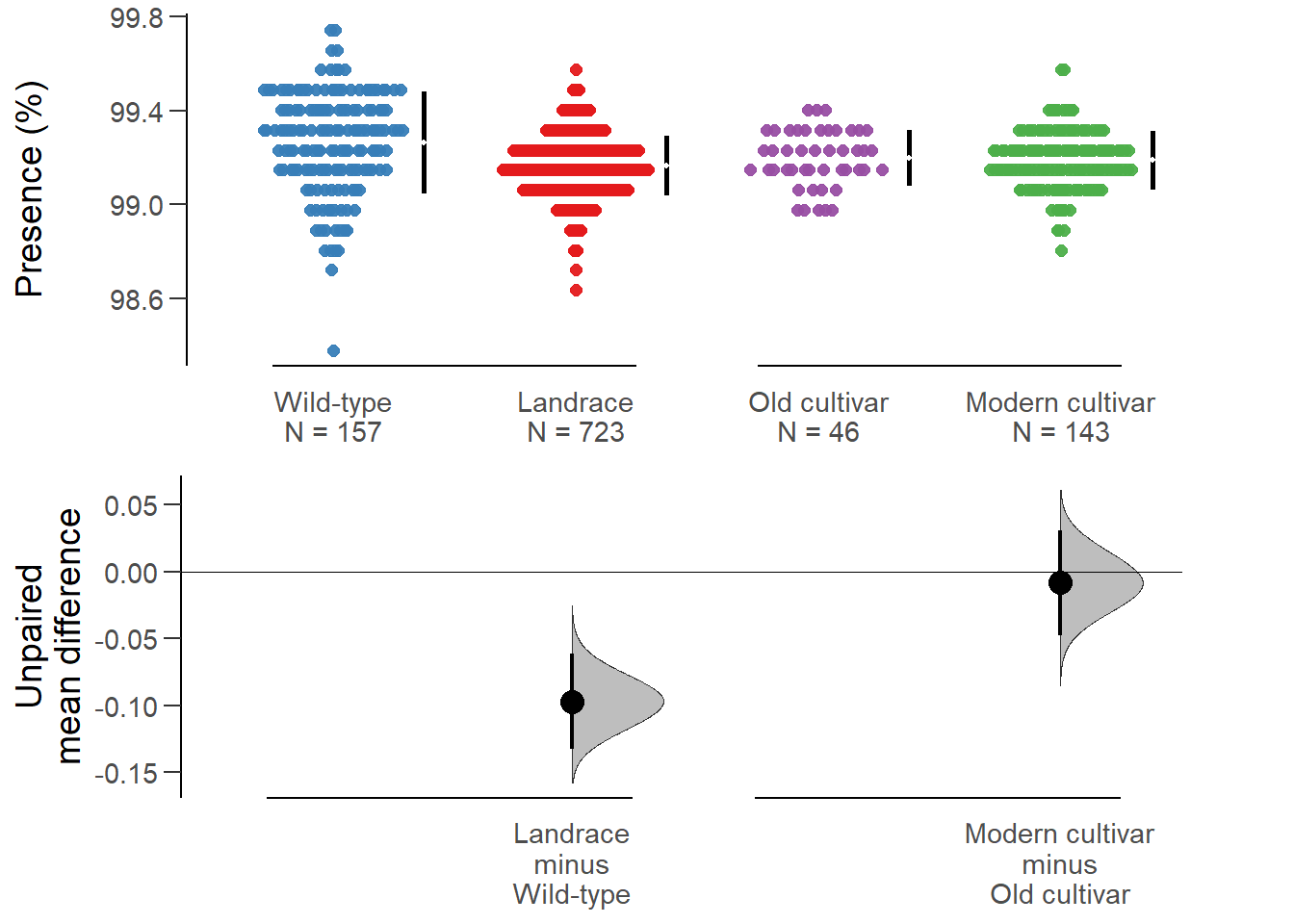
No difference between old and modern cultivars!
RLP
rlp_multi.two.group.unpaired <-
rlp_joined_groups %>% filter(!is.na(`PI-ID`)) %>%
dabest(`Group in violin table`, percs,
idx = list(c("Wild-type", "Landrace"),
c('Old cultivar', 'Modern cultivar')),
paired = FALSE)
rlp_multi.two.group.unpaireddabestr (Data Analysis with Bootstrap Estimation in R) v0.3.0
=============================================================
Good morning!
The current time is 11:04 AM on Friday December 11, 2020.
Dataset : .
The first five rows are:
# A tibble: 5 x 4
names percs `PI-ID` `Group in violin table`
<chr> <dbl> <chr> <fct>
1 AB-01 95 PI458020 Landrace
2 AB-02 95.6 PI603713 Landrace
3 DT2000 95 PI635999 Modern cultivar
4 For 95 PI548645 Modern cultivar
5 HN001 95.6 PI518664 Modern cultivar
X Variable : Group in violin table
Y Variable : percs
Effect sizes(s) will be computed for:
1. Landrace minus Wild-type
2. Modern cultivar minus Old cultivarrlp_multi.two.group.unpaired.meandiff <- mean_diff(rlp_multi.two.group.unpaired)
rlp_multi.two.group.unpaired.meandiffdabestr (Data Analysis with Bootstrap Estimation in R) v0.3.0
=============================================================
Good morning!
The current time is 11:04 AM on Friday December 11, 2020.
Dataset : .
X Variable : Group in violin table
Y Variable : percs
Unpaired mean difference of Landrace (n = 723) minus Wild-type (n = 157)
-0.804 [95CI -0.965; -0.651]
Unpaired mean difference of Modern cultivar (n = 143) minus Old cultivar (n = 46)
-0.0122 [95CI -0.294; 0.265]
5000 bootstrap resamples.
All confidence intervals are bias-corrected and accelerated.plot(rlp_multi.two.group.unpaired.meandiff, color.column=`Group in violin table`,
rawplot.ylabel = 'Presence (%)', show.legend=FALSE)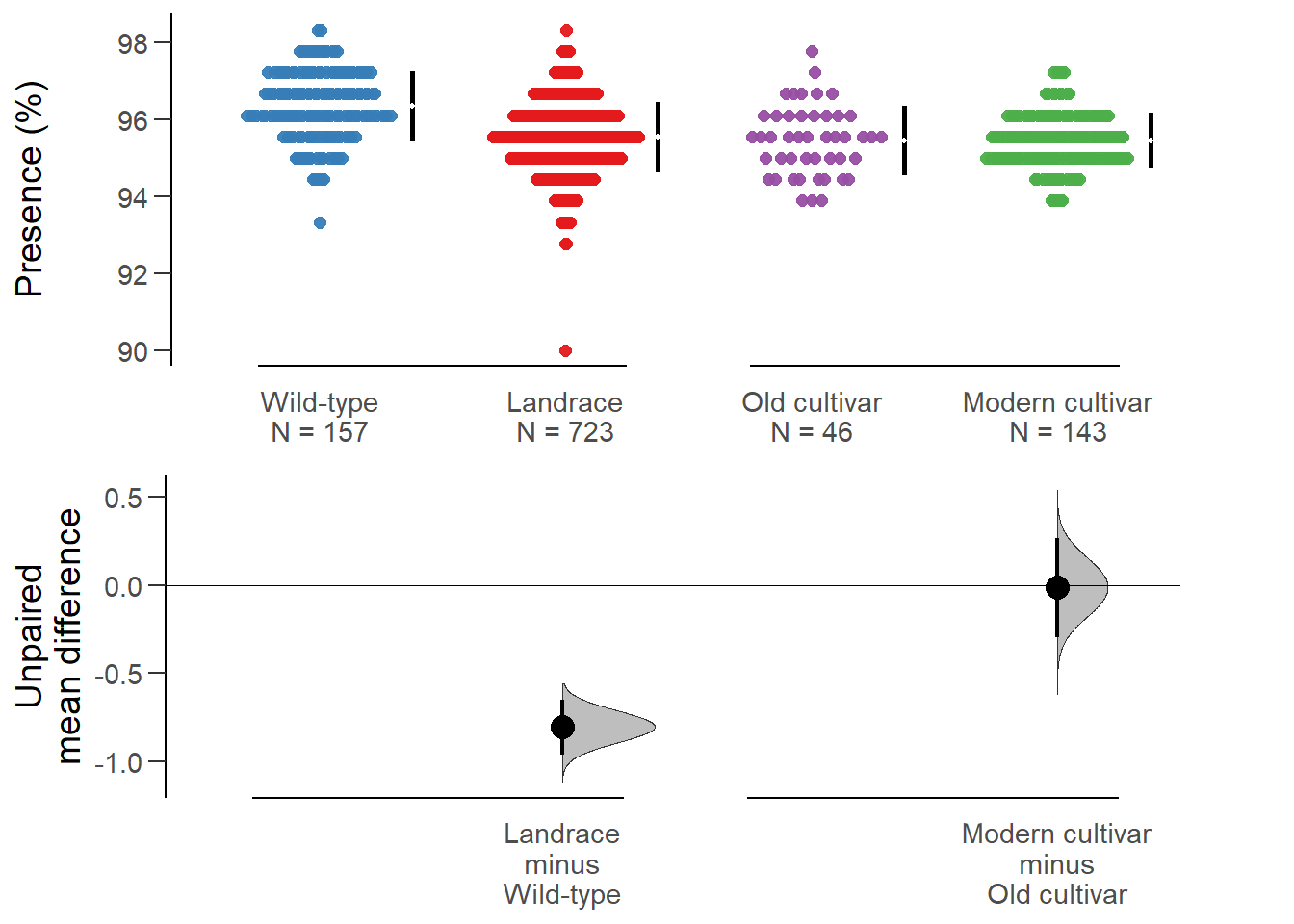
Again, no difference between old and modern cultivars!
Stats - classic t-test
NBS
nbs_joined_groups %>%
filter( !is.na(`PI-ID`) ) %>%
ggplot(aes(x=`Group in violin table`, y = percs,
fill = `Group in violin table`)) +
geom_boxplot() +
scale_fill_manual(values = col_list) +
theme_minimal_hgrid() +
theme(axis.text.x = element_text(size=12),
axis.text.y = element_text(size=12)) +
geom_signif(comparisons = list(c('Wild-type', 'Landrace'),
c('Old cultivar', 'Modern cultivar')),
map_signif_level = T) +
guides(fill=FALSE)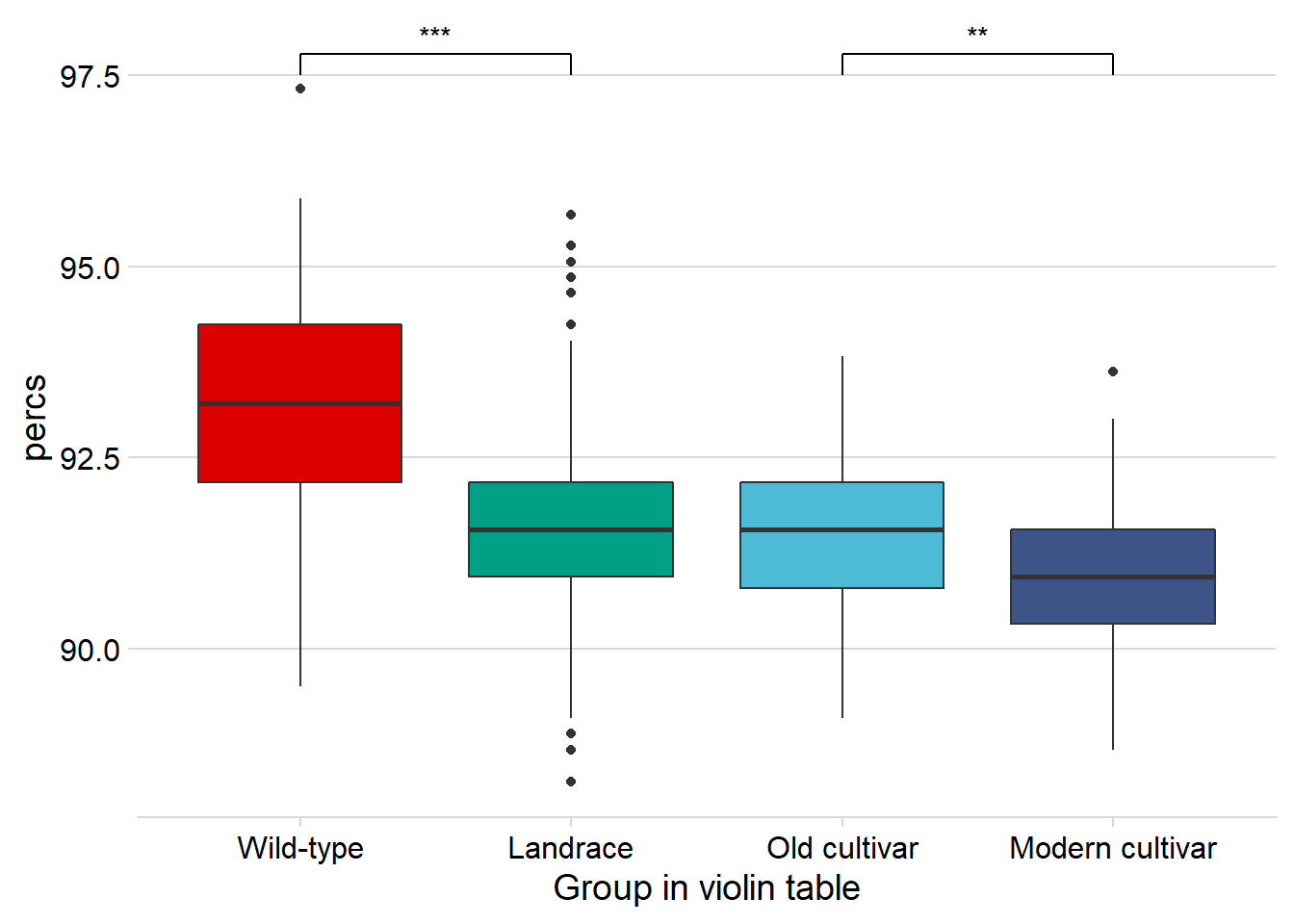
RLP
rlp_joined_groups %>%
filter( !is.na(`PI-ID`) ) %>%
ggplot(aes(x=`Group in violin table`, y = percs,
fill = `Group in violin table`)) +
geom_boxplot() +
scale_fill_manual(values = col_list) +
theme_minimal_hgrid() +
theme(axis.text.x = element_text(size=12),
axis.text.y = element_text(size=12)) +
geom_signif(comparisons = list(c('Wild-type', 'Landrace'),
c('Old cultivar', 'Modern cultivar')),
map_signif_level = T) +
guides(fill=FALSE)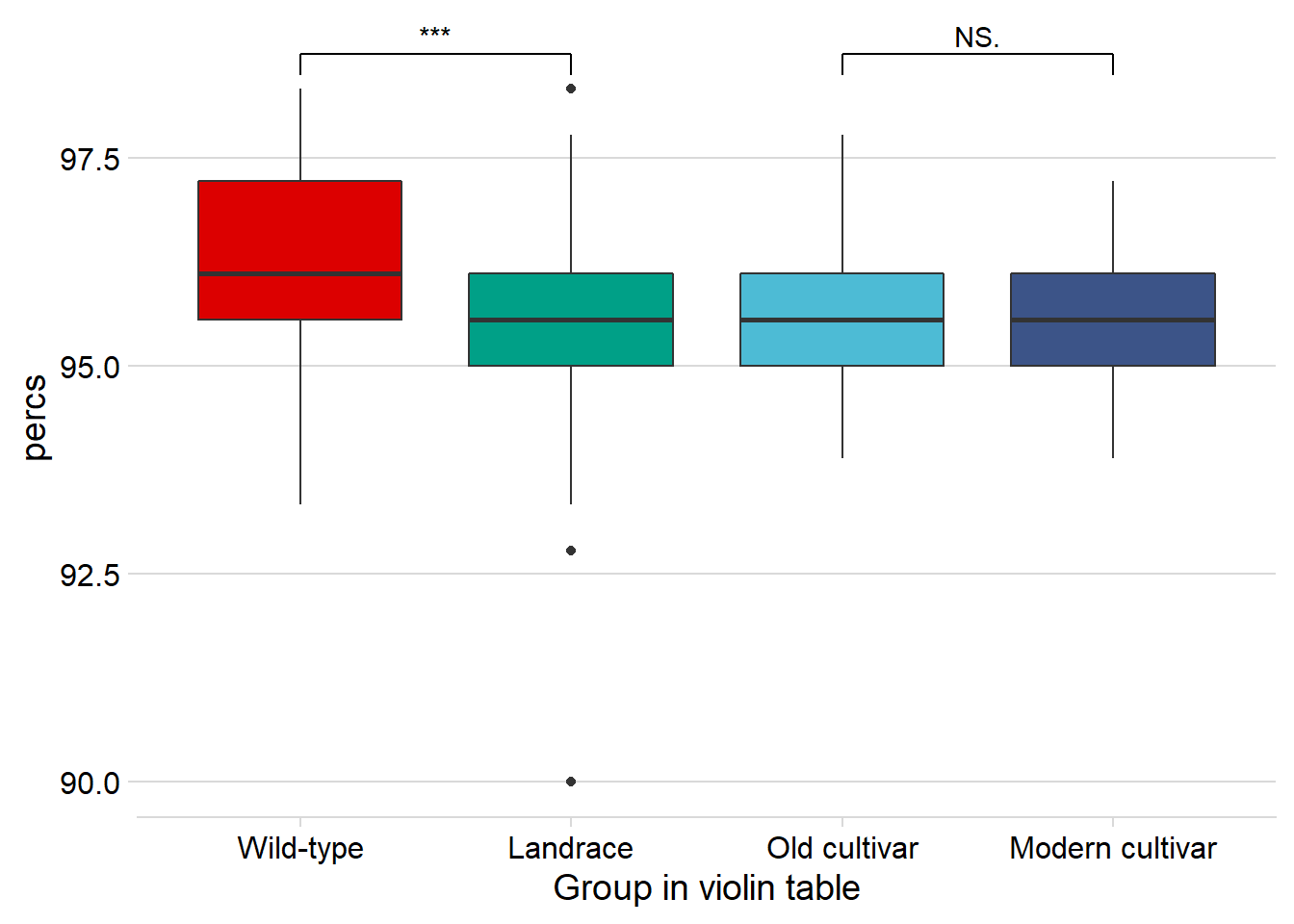
RLK
rlk_joined_groups %>%
filter( !is.na(`PI-ID`) ) %>%
ggplot(aes(x=`Group in violin table`, y = percs,
fill = `Group in violin table`)) +
geom_boxplot() +
scale_fill_manual(values = col_list) +
theme_minimal_hgrid() +
theme(axis.text.x = element_text(size=12),
axis.text.y = element_text(size=12)) +
geom_signif(comparisons = list(c('Wild-type', 'Landrace'),
c('Old cultivar', 'Modern cultivar')),
map_signif_level = T) +
guides(fill=FALSE)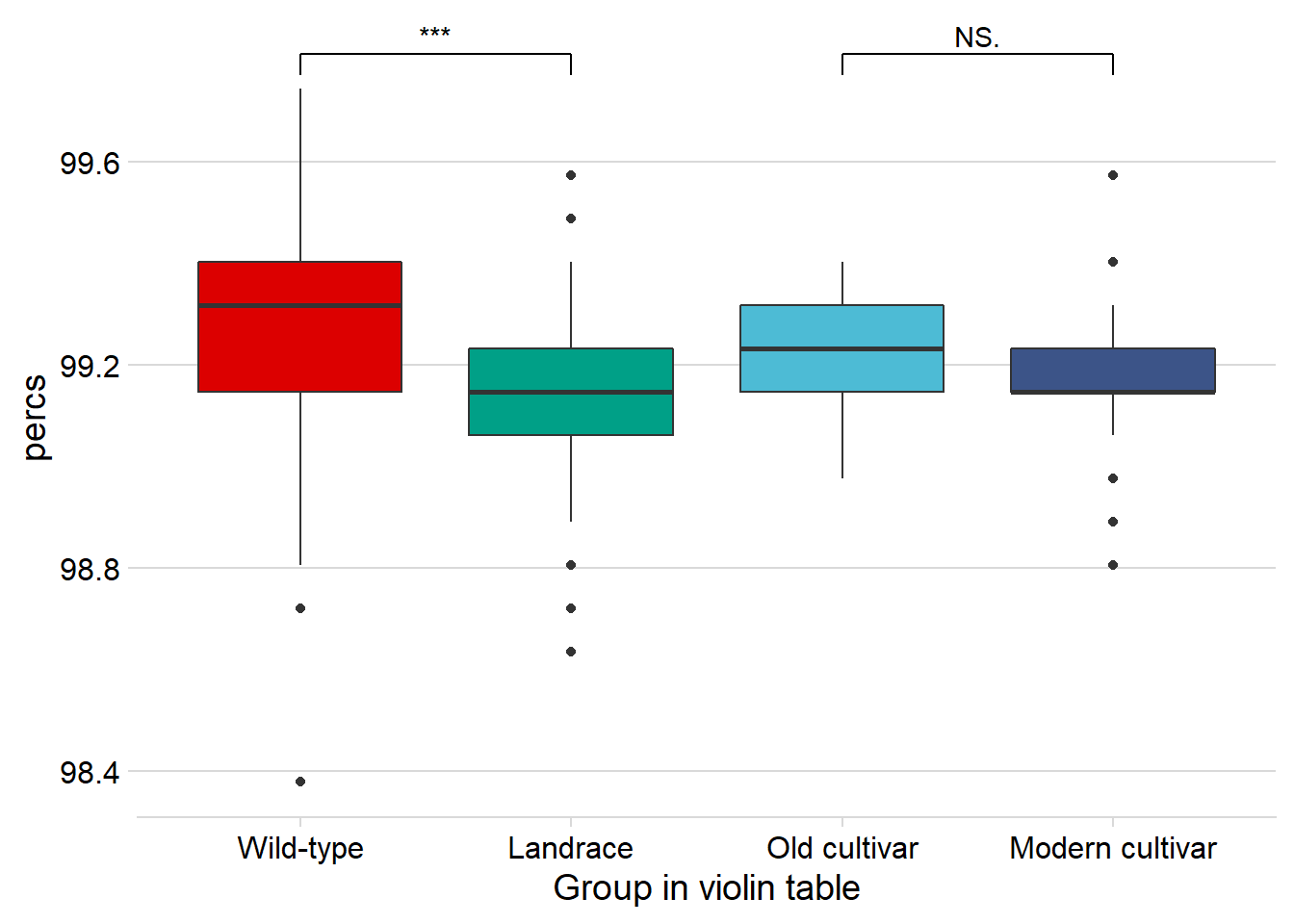
sessionInfo()R version 3.6.3 (2020-02-29)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 17134)
Matrix products: default
locale:
[1] LC_COLLATE=English_Australia.1252 LC_CTYPE=English_Australia.1252
[3] LC_MONETARY=English_Australia.1252 LC_NUMERIC=C
[5] LC_TIME=English_Australia.1252
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] ggforce_0.3.1 ggsignif_0.6.0 cowplot_1.0.0
[4] dabestr_0.3.0 magrittr_1.5 ggsci_2.9
[7] patchwork_1.0.0 forcats_0.5.0 stringr_1.4.0
[10] dplyr_1.0.0 purrr_0.3.4 readr_1.3.1
[13] tidyr_1.1.0 tibble_3.0.2 ggplot2_3.3.2
[16] tidyverse_1.3.0 workflowr_1.6.2.9000
loaded via a namespace (and not attached):
[1] nlme_3.1-148 fs_1.5.0.9000 lubridate_1.7.9 RColorBrewer_1.1-2
[5] httr_1.4.2 rprojroot_1.3-2 tools_3.6.3 backports_1.1.10
[9] utf8_1.1.4 R6_2.4.1 vipor_0.4.5 DBI_1.1.0
[13] mgcv_1.8-31 colorspace_1.4-1 withr_2.2.0 tidyselect_1.1.0
[17] processx_3.4.4 compiler_3.6.3 git2r_0.27.1 cli_2.0.2
[21] rvest_0.3.5 xml2_1.3.2 labeling_0.3 scales_1.1.1
[25] callr_3.4.4 digest_0.6.25 rmarkdown_2.3 pkgconfig_2.0.3
[29] htmltools_0.5.0 dbplyr_1.4.4 highr_0.8 rlang_0.4.7
[33] readxl_1.3.1 rstudioapi_0.11 farver_2.0.3 generics_0.0.2
[37] jsonlite_1.7.1 Matrix_1.2-18 Rcpp_1.0.5 ggbeeswarm_0.6.0
[41] munsell_0.5.0 fansi_0.4.1 lifecycle_0.2.0 stringi_1.5.3
[45] whisker_0.4 yaml_2.2.1 MASS_7.3-51.6 plyr_1.8.6
[49] grid_3.6.3 blob_1.2.1 promises_1.1.1 crayon_1.3.4
[53] lattice_0.20-41 haven_2.3.1 splines_3.6.3 hms_0.5.3
[57] knitr_1.29 ps_1.3.4 pillar_1.4.4 boot_1.3-25
[61] reprex_0.3.0 glue_1.4.2 evaluate_0.14 getPass_0.2-2
[65] modelr_0.1.8 vctrs_0.3.1 tweenr_1.0.1 httpuv_1.5.4
[69] cellranger_1.1.0 gtable_0.3.0 polyclip_1.10-0 assertthat_0.2.1
[73] xfun_0.17 broom_0.5.6 later_1.1.0.1 beeswarm_0.2.3
[77] ellipsis_0.3.1